
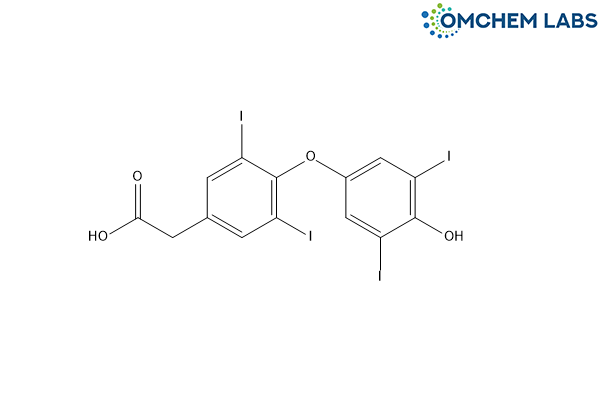
Levothyroxine Impurity D
| Catalogue No |
LEVO-OCL-006 |
| CAS NO |
67-30-1 |
| Molecular Formula | C14H8I4O4 |
| Molecular weight | 747.83 |
| Inquiry Status | In Stock |
| Synonyms | Levothyroxine EP Impurity D T4-Acetic Acid (USP) Tetraiodothyroacetic acid [4-(4-Hydroxy-3,5-diiodophenoxy)-3,5-diiodophenyl]acetic acid |
Detailed Overview of this Impurity: Discover more about Impurity Standard & Analysis
Scientific Overview on the Impurity Profiling of Levothyroxine: Focus on Levothyroxine Impurity D
Introduction
The comprehensive understanding of impurity profiles plays a central role in pharmaceutical development, particularly for active pharmaceutical ingredients (APIs) with narrow therapeutic indices, such as Levothyroxine. The presence of Levothyroxine Impurity D, like any structurally related substance or unintended by-product, demands focused scientific attention to ensure therapeutic safety, product stability, and compliance with regulatory expectations. Impurity profiling is not only a regulatory requirement but a fundamental aspect of drug substance quality management. In the case of Levothyroxine, whose bioavailability is sensitive to minor changes in composition, impurity identification and control becomes critical throughout the product lifecycle.
Formation of Impurities During API Synthesis
Impurities such as Levothyroxine Impurity D can originate through diverse mechanisms during the synthesis and handling of the API. Synthetic routes, reaction intermediates, reagent residues, and process conditions collectively contribute to impurity formation. Slight modifications in parameters like pH, reaction temperature, or solvent purity can result in the development of by-products or incomplete transformations. Additionally, exposure to environmental conditions such as humidity, light, or oxidative stress during storage or packaging may lead to degradation-based impurities. In multistep syntheses, the possibility of carryover from intermediates or residual solvents adds another layer of complexity, emphasizing the importance of understanding the origin and potential behavior of impurities like Impurity D.
Analytical Data Interpretation Techniques
To reliably assess the presence of Levothyroxine Impurity D, robust and sensitive analytical techniques must be implemented. The process typically begins with separation methodologies such as high-performance liquid chromatography (HPLC) or ultra-performance liquid chromatography (UPLC), followed by structural elucidation via spectrometric tools like mass spectrometry (MS) or nuclear magnetic resonance (NMR) spectroscopy. Infrared spectroscopy and ultraviolet detection may also support impurity classification. The interpretive process involves analyzing retention patterns, fragmentation pathways, and spectral data to distinguish impurities from the principal compound. A deep understanding of analytical signatures ensures accurate identification and supports consistent impurity tracking across multiple production batches.
Method Validation for Impurity Detection
To guarantee the reproducibility and reliability of impurity detection methods, comprehensive validation procedures are required. The analytical methodology used for the quantification of Levothyroxine Impurity D must conform to international validation standards. Validation typically includes assessment of method precision, accuracy, specificity, linearity, detection limits, and robustness under varied conditions. These parameters ensure the method’s suitability for routine quality control and regulatory documentation. A validated method also allows early detection of impurities during scale-up or post-approval changes, thus minimizing the risk of undetected impurity spikes in production environments.
Purification Strategies for Reducing Impurities
After impurity identification, strategic purification becomes essential to eliminate or minimize its presence in the final drug substance. Techniques such as recrystallization, liquid-liquid extraction, and preparative chromatography are commonly employed to purify Levothyroxine and reduce levels of Impurity D. Selection of an appropriate purification technique is often based on solubility differences, chemical reactivity, or molecular polarity. Optimizing process parameters during purification not only improves yield and efficiency but also significantly enhances the overall quality of the API. A well-defined purification strategy contributes to consistency and regulatory readiness during commercial production.
Isolation and Characterization of Impurities
When Levothyroxine Impurity D is observed at levels beyond the qualification threshold, isolation and detailed characterization become mandatory. Impurity isolation is typically accomplished using preparative-scale chromatographic techniques. Once isolated, advanced spectroscopic methods are employed to determine the exact structural framework. Mass spectrometry offers insight into molecular weight and fragmentation, while NMR helps in assigning chemical environment and structural conformation. Properly characterized impurities are vital for toxicological evaluations, risk assessment, and justification of impurity limits in regulatory submissions. Establishing impurity reference standards further facilitates batch-to-batch consistency in analytical assessments.
Conclusion
The impurity profiling of Levothyroxine Impurity D encompasses a multidimensional framework involving synthesis pathway review, rigorous analytical evaluation, method validation, impurity control through purification, and comprehensive structural analysis. As pharmaceutical regulations continue to evolve toward greater specificity and quality assurance, maintaining a robust impurity control strategy becomes indispensable. Particularly for APIs like Levothyroxine, where therapeutic consistency is tightly regulated, a well-executed impurity profiling process ensures patient safety, product integrity, and sustained regulatory compliance throughout the product’s commercial lifecycle.