
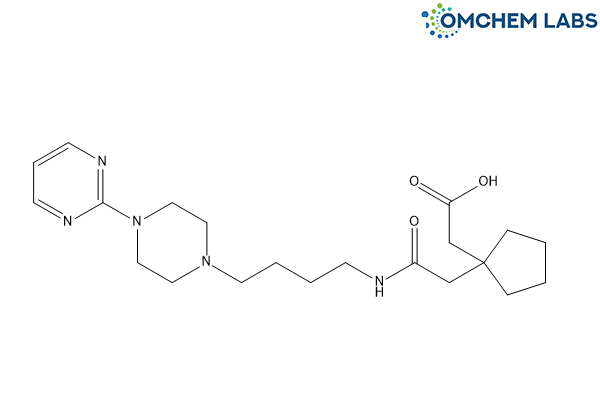
Buspirone Impurity E
| Catalogue No |
BUSP-OCL-008 |
| CAS NO |
257877-43-3 |
| Molecular Formula | C21H33N5O3 |
| Molecular weight | 403.52 |
| Inquiry Status | In Stock |
| Synonyms | [1-[2-Oxo-2-[[4-[4-(pyrimidin-2-yl)piperazin-1-yl]butyl]amino]ethyl] cyclopentyl]acetic acid |
Detailed Overview of this Impurity: Discover more about Impurity Standard & Analysis
Impurity Profiling of Buspirone Impurity E: A Scientific Overview
Introduction
The pharmaceutical industry places strong emphasis on understanding and managing impurities associated with active pharmaceutical ingredients (APIs). Buspirone Impurity E represents a critical area of study, as impurities not only affect therapeutic performance but also influence patient safety, product stability, and regulatory compliance. Comprehensive profiling of such impurities forms the backbone of quality control and ensures adherence to international guidelines. Evaluating the chemical origins, analytical detection methods, and strategies for mitigation allows researchers and manufacturers to establish robust quality frameworks throughout the drug development lifecycle.
Formation of Impurities During API Synthesis
The occurrence of impurities such as Buspirone Impurity E is closely tied to the complexities of synthetic chemistry. Throughout multi-step synthesis, unwanted chemical entities can be generated due to incomplete reactions, rearrangements, degradation pathways, or interaction of intermediates with solvents, catalysts, or reagents. Post-synthetic instability, influenced by environmental factors such as light exposure, oxidative stress, and temperature variation, may also contribute to impurity development. Recognizing these potential sources provides the foundation for designing effective impurity control strategies, ensuring the desired purity profile of the final drug substance.
Analytical Data Interpretation Techniques
Accurate profiling of Buspirone Impurity E relies on modern analytical sciences. Chromatographic techniques, including high-performance liquid chromatography (HPLC) and gas chromatography (GC), are central to impurity monitoring, offering both separation and quantification capabilities. Coupled with spectroscopic and spectrometric methods such as mass spectrometry (MS), nuclear magnetic resonance (NMR), and infrared (IR) analysis, a comprehensive understanding of impurity composition can be achieved. Interpretation of chromatograms, spectral peaks, and fragmentation patterns allows researchers to distinguish between structurally related components and provides insights into impurity origin and behavior. The integration of these methods ensures high confidence in the accuracy of profiling.
Method Validation for Impurity Detection
Before impurity data can be considered reliable, the underlying analytical methods require systematic validation. In the case of Buspirone Impurity E, method validation serves to confirm that chosen techniques deliver reproducible, accurate, and sensitive outcomes. Validation parameters include specificity, accuracy, precision, linearity, robustness, and detection capability at trace levels. Compliance with international standards such as those outlined by the International Council for Harmonisation (ICH) provides assurance that analytical procedures are scientifically sound and regulatory-ready. Rigorous validation safeguards the reliability of results, ensuring that impurities are consistently identified and quantified under defined conditions.
Purification Strategies for Reducing Impurities
Effective purification plays a central role in reducing the presence of Buspirone Impurity E and related contaminants. Depending on the chemical characteristics of both the impurity and the target molecule, several approaches may be employed. Crystallization, solvent extraction, and chromatographic purification are frequently applied, each offering unique advantages in selectivity and yield. Distillation and phase separation may also be useful for volatile or solvent-based impurities. A carefully optimized purification plan ensures that the final API meets pharmacopeial standards, while minimizing product loss and process inefficiencies. The strategic integration of purification within manufacturing helps maintain consistent quality and safety.
Isolation and Characterization of Impurities
When impurities such as Buspirone Impurity E exceed reporting thresholds or remain structurally undefined, isolation becomes a critical step. Advanced preparative chromatography and separation techniques allow for recovery of sufficient impurity quantities for detailed analysis. Once isolated, structural elucidation is typically performed using NMR, MS, and complementary spectroscopic tools. The goal is to confirm the molecular architecture and assess potential pharmacological or toxicological significance. These insights enable the establishment of appropriate impurity limits and support risk assessment within regulatory submissions. Developing a reference standard for recurring impurities further enhances routine analytical control.
Conclusion
Impurity profiling of Buspirone Impurity E illustrates the essential intersection of chemical understanding, analytical science, and regulatory compliance within pharmaceutical development. From identifying the conditions under which impurities arise, to applying validated analytical methods, and implementing purification and isolation techniques, each stage contributes to safeguarding drug quality and patient safety. A well-structured impurity control strategy not only fulfills regulatory obligations but also enhances confidence in the therapeutic consistency of the API. As drug development advances, continued focus on impurity profiling remains fundamental to pharmaceutical excellence and public health protection.