
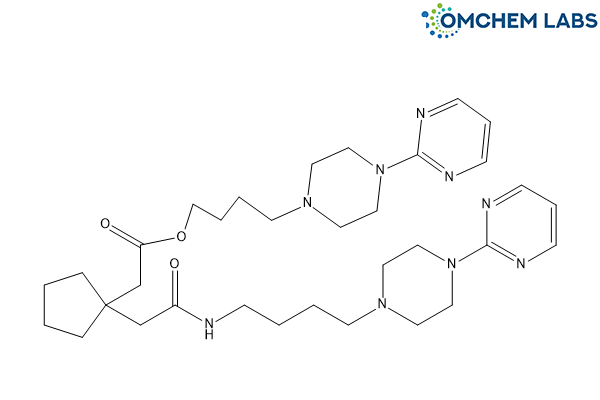
Buspirone Impurity F
| Catalogue No |
BUSP-OCL-009 |
| CAS NO |
2512210-24-9 |
| Molecular Formula | C33H51N9O3 |
| Molecular weight | 621.82 |
| Inquiry Status | In Stock |
| Synonyms | Buspirone Open-Ring Dimer Impurity |
Detailed Overview of this Impurity: Discover more about Impurity Standard & Analysis
Impurity Profiling of Buspirone Impurity F: A Scientific Perspective
Introduction
In pharmaceutical sciences, impurity profiling has evolved into a cornerstone of quality assurance, ensuring that active pharmaceutical ingredients (APIs) remain safe, effective, and consistent throughout their lifecycle. The presence of even trace-level impurities can alter therapeutic efficacy or raise safety concerns, making a systematic study of impurity formation, identification, and control essential. Buspirone Impurity F represents one such related compound of interest, requiring comprehensive evaluation to align with regulatory expectations and scientific standards. By integrating analytical chemistry, process understanding, and regulatory compliance, impurity profiling of Buspirone Impurity F ensures the integrity of the final product and safeguards patient safety.
Formation of Impurities During API Synthesis
The appearance of impurities in Buspirone Impurity F is often linked to complexities inherent in multi-step organic synthesis. Several contributing factors can be recognized: unreacted starting substances, rearranged intermediates, catalyst residues, and adventitious by-products from side reactions. Environmental conditions during synthesis, such as elevated temperature, prolonged reaction times, or variations in pH, may also promote unwanted degradation. Beyond synthetic origins, impurities can emerge during storage and handling, as APIs are frequently sensitive to moisture, light, and oxidative stress. Understanding these mechanistic pathways helps to anticipate impurity formation and guides the design of more robust manufacturing processes.
Analytical Data Interpretation Techniques
Accurate impurity profiling of Buspirone Impurity F requires sophisticated instrumentation capable of detecting, differentiating, and characterizing chemically similar species. Chromatographic platforms like high-performance liquid chromatography (HPLC) and gas chromatography (GC) provide resolution and quantification, while hyphenated systems such as LC–MS and GC–MS supply additional structural insight. Spectroscopic methods, particularly NMR and IR, assist in confirming chemical identity. Interpretation of data involves evaluating chromatographic peaks, spectral fingerprints, and fragmentation pathways to distinguish known impurities from novel or unexpected entities. This interpretive process ensures not only compliance with pharmacopeial monographs but also supports scientific understanding of the compound’s behavior.
Method Validation for Impurity Detection
Reliability of impurity analysis depends on method validation that demonstrates the robustness and reproducibility of the chosen techniques. For Buspirone Impurity F, validation covers a spectrum of parameters, including accuracy, precision, linearity, specificity, sensitivity, and reproducibility, as defined by regulatory guidelines such as ICH Q2. Validated methods confirm that impurities can be consistently detected across different laboratories and conditions, avoiding false negatives or misidentification. This step is crucial not only for regulatory approval but also for establishing long-term confidence in impurity monitoring during both development and commercial manufacturing.
Purification Strategies for Reducing Impurities
Purification serves as a vital step in ensuring that Buspirone Impurity F and related substances are controlled within acceptable thresholds. Depending on the physicochemical nature of the compound, approaches such as crystallization, recrystallization, solvent extraction, and preparative chromatography may be applied. Each strategy is selected on the basis of efficiency in separating structurally similar molecules while maintaining the chemical stability of the target API. A systematic purification plan allows manufacturers to minimize residual impurities while optimizing yield and process economics, ensuring that the final drug substance conforms to quality specifications.
Isolation and Characterization of Impurities
Once an impurity such as Buspirone Impurity F is identified, further steps are often required to isolate it for structural confirmation and toxicological assessment. Techniques such as preparative HPLC or flash chromatography allow separation at sufficient scale, followed by in-depth characterization using advanced spectroscopic and spectrometric methods. These analyses provide molecular-level detail on chemical identity, enabling researchers to propose mechanisms of formation and establish potential impact on drug safety. Isolated impurities may also serve as reference materials for ongoing analytical studies, strengthening the foundation of impurity profiling programs.
Conclusion
The profiling of Buspirone Impurity F illustrates the interdisciplinary nature of impurity science, encompassing synthesis, analytical chemistry, validation, purification, and characterization. By integrating these facets, pharmaceutical scientists can develop comprehensive impurity control strategies that not only satisfy regulatory expectations but also contribute to patient safety and therapeutic reliability. In a broader sense, impurity profiling reflects the pharmaceutical industry’s commitment to scientific rigor, product consistency, and the continual refinement of drug quality standards.