
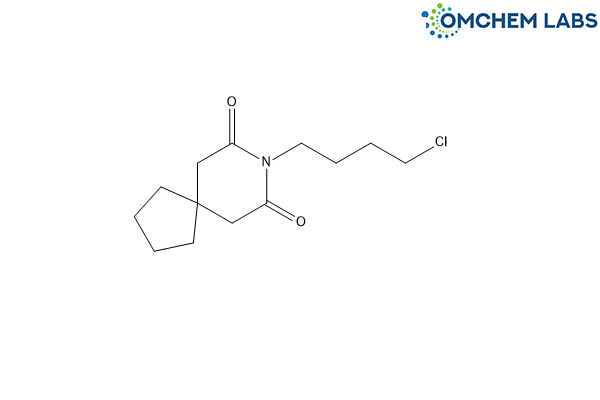
Buspirone Impurity L
| Catalogue No |
BUSP-OCL-013 |
| CAS NO |
21098-11-3 |
| Molecular Formula | C13H20ClNO2 |
| Molecular weight | 257.76 |
| Inquiry Status | In Stock |
| Synonyms | 8-(4-Chlorobutyl)-8-azaspiro[4.5]decane-7,9-dione |
Detailed Overview of this Impurity: Discover more about Impurity Standard & Analysis
Impurity Profiling of Buspirone Impurity L: A Scientific Perspective
Introduction
The pharmaceutical industry places significant emphasis on the identification and control of impurities in active pharmaceutical ingredients (APIs). Buspirone Impurity L represents one such entity requiring careful evaluation to maintain the safety and efficacy of the parent drug. Impurities may compromise therapeutic outcomes or increase toxicological risks, making their detection, quantification, and elimination essential. The study of impurity profiling is not merely a regulatory requirement; it is also an integral aspect of ensuring consistent drug quality and patient safety.
Formation of Impurities During API Synthesis
The synthetic pathway of an API is often complex and prone to generating unintended chemical species. In the case of Buspirone Impurity L, potential origins include incomplete reactions, transformation of intermediates, rearrangements of unstable compounds, or the persistence of residual solvents and catalysts. Reaction conditions such as temperature, pH, solvent polarity, and time of exposure can all influence the impurity landscape. Beyond the laboratory process, storage conditions—light, humidity, and oxidation—may also play a role in producing degradation-related impurities. Understanding these mechanisms provides valuable insight into controlling impurity formation early in the development process.
Analytical Data Interpretation Techniques
The accurate identification of impurities relies heavily on modern analytical platforms. For Buspirone Impurity L, tools such as high-performance liquid chromatography (HPLC), gas chromatography (GC), mass spectrometry (MS), and nuclear magnetic resonance (NMR) are commonly employed. Each technique offers complementary information: chromatographic retention times for separation, mass spectra for molecular weight and fragmentation patterns, and NMR for structural elucidation. Data interpretation requires careful correlation of these results to confirm impurity identity and monitor trends across production batches. The ability to interpret data effectively ensures compliance with pharmacopeial guidelines and supports ongoing quality control.
Method Validation for Impurity Detection
The credibility of impurity profiling depends on the robustness of the analytical methods applied. For Buspirone Impurity L, method validation is performed under internationally accepted frameworks such as ICH Q2(R1). Critical attributes evaluated include accuracy, precision, specificity, sensitivity, linearity, reproducibility, and robustness. A validated method assures that impurities can be detected reliably, even at trace levels, and that results remain consistent across laboratories and manufacturing sites. Such validation safeguards regulatory acceptance and provides assurance of quality for both developers and patients.
Purification Strategies for Reducing Impurities
The removal or reduction of impurities requires purification techniques tailored to the properties of both the API and the impurity. Strategies applicable to Buspirone Impurity L include crystallization, which leverages solubility differences, distillation for volatile components, solvent partitioning for selective extraction, and advanced chromatographic approaches for fine resolution. An effective purification plan enhances the purity of the final drug substance without compromising yield or stability. Selecting the optimal approach often depends on the chemical behavior of the impurity relative to the desired compound.
Isolation and Characterization of Impurities
When impurities such as Buspirone Impurity L exceed predefined thresholds or remain unidentified, isolation becomes necessary. Techniques such as preparative chromatography or specialized extraction methods enable the separation of impurities in sufficient quantity for structural studies. Characterization follows through advanced spectroscopic and analytical tools, including MS, NMR, and IR. Structural elucidation not only confirms the impurity’s chemical identity but also aids in evaluating potential toxicological risks. These insights are essential for establishing impurity specifications and regulatory submissions.
Conclusion
The impurity profiling of Buspirone Impurity L highlights the multifaceted approach required to ensure the safety, quality, and regulatory compliance of pharmaceutical APIs. From understanding impurity formation during synthesis, to employing robust analytical and validated detection methods, to implementing efficient purification and characterization strategies, each step contributes to a comprehensive impurity control framework. By addressing impurities proactively, the pharmaceutical industry ensures not only product consistency but also the trust and well-being of patients worldwide.